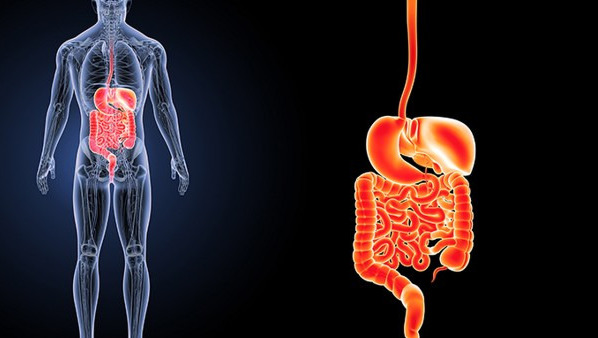
薄基底膜肾病可能会遗传,通常属于常染色体显性遗传模式。薄基底膜肾病是一种以肾小球基底膜变薄为特征的良性肾脏疾病,多数患者无明显症状或仅表现为镜下血尿。
薄基底膜肾病的遗传性主要与COL4A3或COL4A4基因突变有关。这些基因编码IV型胶原蛋白,是肾小球基底膜的重要组成成分。当基因发生突变时,可能导致基底膜结构异常变薄。家族中若存在该病患者,直系亲属患病概率可能增加。多数患者病情稳定,肾功能长期保持正常,仅需定期监测尿常规和肾功能。
少数情况下薄基底膜肾病可能与其他遗传性肾病如Alport综合征重叠,此时可能伴随进行性肾功能减退或感音神经性耳聋等症状。基因检测可帮助鉴别诊断,尤其对于有家族史或合并其他异常表现的患者。建议有生育需求的患者进行遗传咨询,必要时通过产前诊断评估胎儿风险。
薄基底膜肾病患者应避免剧烈运动和肾毒性药物,每年至少进行一次尿检和肾功能检查。若出现蛋白尿或血压升高,需及时就医干预。日常保持低盐优质蛋白饮食,控制水分摄入,有助于减轻肾脏负担。