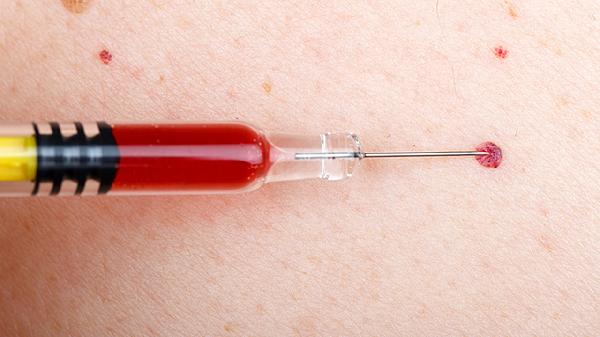
血友病甲主要由遗传性凝血因子Ⅷ缺乏引起,属于X染色体连锁隐性遗传病,可能由基因突变、家族遗传、自发突变、获得性抗体等因素导致。
约70%患者有家族遗传史,因F8基因突变导致凝血因子Ⅷ合成障碍。建议定期监测凝血功能,可输注重组人凝血因子Ⅷ浓缩剂、冷沉淀或新鲜冰冻血浆。
30%患者为自发突变,常见于F8基因内含子22倒位。需避免创伤性活动,急性出血时使用去氨加压素、凝血酶原复合物或艾美赛珠单抗。
部分患者体内产生抗凝血因子Ⅷ抗体,导致继发性凝血功能障碍。需免疫抑制治疗,药物可选利妥昔单抗、环磷酰胺联合糖皮质激素。
合并血管性血友病时加重出血倾向,可能与血管性血友病因子缺陷有关。需联合输注含VWF的凝血因子浓缩剂,避免使用阿司匹林等抗血小板药物。
患者应避免剧烈运动和外伤,均衡摄入富含维生素K的菠菜、西蓝花等食物,出血发作时立即就医处理。